
As someone considering participating in a clinical trial, it’s natural to worry about potential side effects or other serious issues arising while in a trial. While like any treatment option there are inherent risks for participating in a study, it's essential to understand both the risks and how policies keep patient safety a top priority in clinical trials.
New treatments have to pass many stages before they get to clinical trials
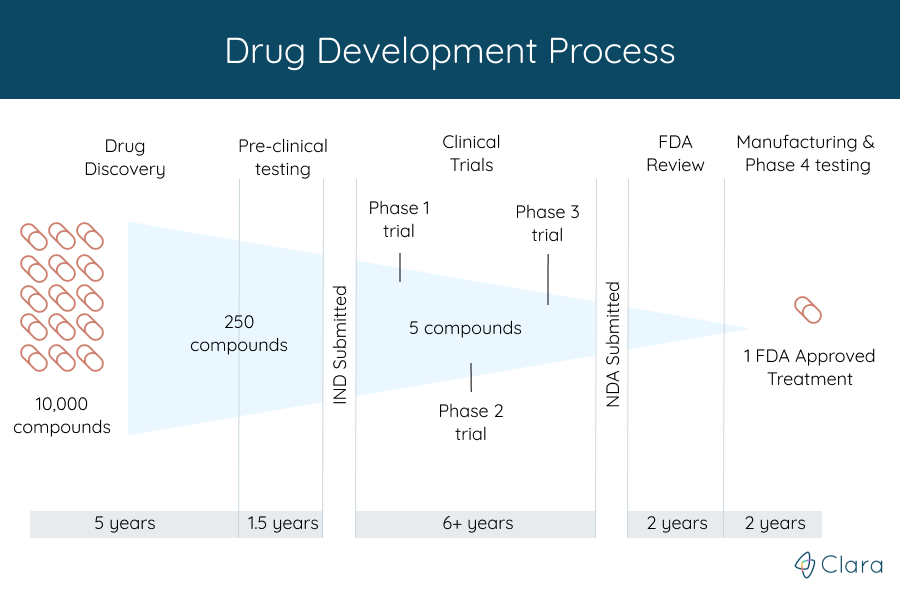
Clinical trials are the last step in the 10-15 year research process to develop a new treatment for a condition. Before starting a clinical trial for a new drug, for example, the treatment first had to be:
- Discovered or created, which can take over 5 years
- Successfully tested in labs cell studies. These are usually the first tests done on a new treatment. To see if it might work, researchers watch the effects of the new treatment on animal or human cells that are grown in a lab.
- Tested in animal studies. Treatments that look encouraging in cell studies are tested next in live animals. This gives researchers a better idea of how safe the new treatment is. The testing process can take around 1.5 years in total.
Permission from the FDA
After medical experts have shown in both laboratory and in animal research that a particular treatment has a good chance of being better than what's currently available for people with a specific disease, they have to get permission from the FDA to start a clinical trial. The FDA, or Food and Drug Administration, is a federal institution that protects participants throughout the clinical trial process by working closely with researchers to make sure that trials are safe.
One of the main ways they accomplish this goal is by only allowing well-studied treatments to become enrolling clinical trials. To get permission from the FDA, the organization in charge of the study, called the sponsor, submits an Investigational New Drug (IND) application. This application combines all of the data on the treatment to prove they have done the needed steps before starting a clinical trial. It includes information about:
Pre-clinical studies: Results from the cell and animal studies, which let the FDA decide whether the treatment is safe to move on in a clinical trial with humans. This may also include any experience with the treatment in people, as some treatments are studied in other countries before entering clinical trials in the United States.
Manufacturing information: How the treatment is made, exactly what's in it, who makes, it, and the different qualities. This helps the FDA make sure that the company can consistently make the treatment in the same way.
Protocol for the clinical trial: Study protocols, which are detailed plans for the planned clinical trials, are evaluated to see if the study exposes patients to any unnecessary risks. The protocol includes:
- The reason for doing the study
- Rules about who can participate (eligibility criteria)
- The number of people needed for the trial
- Details of the treatment and safety information
- The specifics around tests, evaluations, and information that will be collected during the study.
Information on the medical professionals who will supervise the study is also reviewed to make sure they're qualified. Lastly, the sponsor must commit to following all the policies around studying a new treatment, and having the study reviewed by an institutional review board.
Institutional Review Board Approval
In addition to the FDA's approval, before a clinical trial can be started, a panel of scientific and medical experts must decide whether or not it’s ethical to ask patients to volunteer for the experimental treatment. Institutional review boards (IRBs), are groups of professionals with diverse experiences in research, medicine, and ethics that review and approve clinical trials before they are started. The IRB review is an essential step to ensuring that patients best interests are kept at the core of clinical trials, and their safety is well protected.
Without IRB approval, a clinical trial cannot begin. IRBs withhold approval from a trial as long as they have any concerns about patient safety and trial structure, and make sure that the study has been designed, as much as possible, to make sure the people in it will be safe.
Of the many potential treatments tested in early stages, very few are determined to be promising enough to be in a clinical trial.
Who monitors safety during the trial?
There are state, national, and international regulations and policies in place to protect the rights and safety of people during and before they are even enrolled in a clinical trial. These laws ensure that clinical trials follow strict scientific and ethical principles. In the United States, both the Office of Human Research Protections and FDA oversee clinical studies to protect participants. They ensure compliance with their regulations by conducting regular on-site inspections. In extreme cases where the FDA feels patients are not being properly protected, they can and will shut down the study.
Clinical trials are run by medical professionals, who are required to follow strict rules enforced by the federal government to make sure that participants are safe, and the protocol is being followed. Each trial includes doctors and nurses with specific responsibilities that allow the clinical trial teams at every location to uphold the same standard of care. Some roles and responsibilities include:
Principal investigator
The principal investigator (PI) of a clinical trial is a medical expert in both the condition and treatment being studied. He or she is responsible for supervising all aspects of the trial to ensure the trial is safe, following the protocol, and compliant with all regulations.
Study coordinator
The study coordinator is responsible for day-to-day operations of the clinical trial, and is typically the first person potential participants interact with on the trial team. Often, this role is filled by a nurse or another individual with a clinical background. The study coordinator helps explain basic details of the study to patients, and works with the rest of the study team to answer any questions they may have. The FDA works to make sure that the way coordinators are presenting their trials to patients is in an accurate and transparent way.
Other physicians and nurses
Certified physicians and nurses who work at the medical center will provide care to patients during a trial. They manage a patient's general health and treat patients according to the study’s protocol. They also work with the principal investigator to report how patients are faring on the treatment.
Study participants receive careful medical attention and are closely observed for potential safety concerns. The process for documenting potential side effects varies trial to trial, but all serious problems will immediately be reported to the medical team. To ensure safety and transparency throughout the entire study, the study team is required to immediately tell participants any new risks, benefits, or side effects they find during the trial.
Throughout the study, the IRB that reviewed the study will also continue to work closely with the principal investigator to ensure safety continues to be a top priority. Along with the IRB, most clinical trials are closely supervised by a Data and Safety Monitoring Committee. The Committee, made up of experts in the condition the trial is treating, regularly monitor what's happening throughout the study. If they believe that the trial treatment is not working or is harming participants, they will stop the trial right away.
Understanding the risks
Many trial participants are accessing brand new approaches to treatment, that can possibly involve serious side effects. This is why it’s required that patients and families be informed of all potential risks before they are allowed to participate - in an unbiased, easy to understand way. This is known as informed consent.
Informed consent includes a thorough explanation of all necessary information, with time provided for the patient to ask questions and discuss the topic with loved ones. Formal voluntary consent must be obtained before enrollment is allowed, and researchers are required to continually educate the patient through the entire duration of the study.
Taking the time to review the informed consent form (ICF) for the study, which gives patients important information to understand whether or not they want to participate, is one of the most important parts of the process.
Informed consent forms explain:
-
The goal of the study
-
Details about the treatment being tested
-
Any potential risks
-
The study design (Will everyone receive the study treatment? Will anyone receive a placebo? Will I know which treatment I'm on?)
-
An outline of all the visits and procedures involved
-
Information on all potential financial costs
-
Payment and reimbursement instructions
-
A confidentiality statement
The ICF is regulated to ensure that the information is accurately represented, holds researchers accountable for negligence, and protects the rights of participants. Much of this process will take place during the first in person visit for a trial, which is why the initial appointment is usually longer than the others. If English is not your preferred language, be sure to inform the researcher, who should provide you an alternative way of understanding the form.
After reading your ICF, you should feel well versed in the purpose of the study, how the trial will be run, all of the potential risks, and what your role will be.
You can leave the trial at any time
The most important thing to keep in mind is that participation in a trial is always voluntary. Even after enrolling, patients always have the right to leave the study at any time. Participants can also refuse particular treatments or tests within a trial, although it may make you ineligible to continue the study.
Participants can leave for any number of reasons, even a simple change of mind. The most common are:
-
The treatment isn’t working for the patient
-
The patient experiences serious side effects during the study
Keep in mind that patient participation is always highly valued -- even early termination of a clinical trial can provide important medical information to the researcher.
If you're considering participating in a trial
If you're thinking about participating in a clinical trial, it's also important to know that every treatment goes through four phases of testing. Each phase has a different purpose, and helps doctors learn more about the treatment being studied.
Although the specific treatment’s safety and efficacy is monitored throughout each phase, what phase that a clinical trial is in can be a factor to consider when finding and deciding between different studies you might want to participate in because it gives you an idea of how much information is currently known about it. For example, a treatment in a phase 3 study has already been through studies in people from the earlier phases that were run, and has more safety information than a treatment that's still in phase 1 testing. There are also phase 4 trials that involve treatments which have already been approved by the FDA.
Regardless of what phase it's in, every clinical trial has to follow the strict regulations about protecting patient safety to make sure every participant stays safe.



